中国肿瘤药物创新,进入效率为王时期 | 海斌访谈
今天的中国肿瘤药物创新,是历史上不曾有过的。
药审改革促成了资源向创新领域集中,这有别于仿制药主导的时期。而肿瘤药物的开发,吸引了最多的医疗资源。
中国是全球肿瘤患者最多的国家,而且发病率还有上升趋势。针对肿瘤药物,研发生态在日趋完善,一些本土的创新成果也已经出现。同时,医药创新企业面临的竞争也是高烈度的:研发或商业化环节的落后,都可能拖垮一家创新药企。
共识
跨国药企和本土药企的高管们有一个共识:中国市场的新药上市速度在加快,竞争烈度在加强。
“过去在大的药企,每个药不做四五年的临床试验是进不了中国市场的,中国病人还要再等四五年药物才能进医保”,基石药业大中华区总经理赵萍对第一财经记者表示,“现在我们的普拉替尼和美国同时开始临床实验,美国获批的时间仅比国内早六个月”。普拉替尼是基石药业开发的一款RET抑制剂,用于既往接受过含铂化疗的RET基因融合阳性的局部晚期或转移性非小细胞肺癌。
赵萍已经在医药企业摸爬滚打二十多年时间,在加入基石药业之前,她是百时美施贵宝的中国区总经理。在百时美施贵宝期间,她推动了全球首个PD-1抑制剂欧狄沃在中国上市。“审批制度的改革,加速了一些药物的批准速度。”她评价说。
赵萍的继任者,百时美施贵宝的现任中国总经理陈思渊持有类似的观点。
去年的三月份,百时美施贵宝的欧狄沃获得中国药品监督管理局的批准,联合含氟尿嘧啶和铂类药物化疗用于一线治疗晚期或转移性胃癌、胃食管连接部癌或食管腺癌患者。欧狄沃由此成为首款在中国拥有胃癌适应症的PD-1抑制剂。
“在中国全面深化药品审批制度改革的背景下,此次的中国获批距离该适应症获得美国食品药品监督管理局批准仅隔了4个半月”,陈思渊此前对第一财经记者表示。
这种共识产生的背景是,中国的药物审核制度改革已经进行了六年时间。
中国药物审评改革开始于2015年,当时国务院印发的《关于改革药品医疗器械审评审批制度的意见》释放出清晰的信号。此后陆续出台临床默示许可期等一系列政策,从制度上逐步完善了医药产业创新的顶层设计。
从2015年至今大约6年时间,这对于创新药物而言,通常不足以完成研发上市的全生命周期。不过,中国医药创新促进会、中国外商投资企业协会药品研制和开发行业委员会共同发布的研究报告《构建中国药物创新生态体系》认为,目前“中国建成了一个相对完整的医药创新生态系统”,改革激发了本土和跨国药企、生物科技公司、投资者、学术界、监管等各方的良性互动。
中国既是全球第二大的医药市场,也是全球肿瘤患者最多的国家,对于肿瘤药物的临床需求迫切。肿瘤药物领域获得的资源投入,尤其引人注目。单从临床数据来看, 2019年中国在肿瘤的临床研究544个,2020年达到711个,2021年截至8月份已经达到了535个。
“未来5年百时美施贵宝将有近30个新产品和适应症引入”,陈思渊透露,该公司准备在2025年将目前1600人左右的员工数量增加一倍,而扩张开始于研发团队,以确保新管线的布局、临床试验以及注册方面工作的开展。
“活跃的商业环境整体是好事情,但这也需要厂家有竞争力。在医药领域,特别是生物医药领域,竞争力是通过研发投入来实现的。”陈思渊对第一财经记者表示。
而研发,对于中国本土的创新药企不是容易的事。
竞争临床资源
肿瘤药物的临床阶段投入普遍要高于其他药物。
“无论是投入的时间周期,还是整体的投入费用方面”,药研社首席医学官陈祖武对第一财经记者表示,肿瘤药物的研发都更有挑战性,比如同样的临床三期研究,肿瘤药物的投入可能达到心血管药物的两倍。
2017年的6月,中国正式加入ICH(人用药品注册技术要求国际协调会)。这是中国与发达国家药物开发标准的进一步接轨,提高了国内制药企业研发生产的规范性。
此后,监管机构对于临床阶段又进行了两项重要的改革:将临床试验申请由审批制改为到期默认制,临床试验机构资格认定实行备案制。
新监管框架改善了临床研究的速度和流程。
“在这个框架下,我们临床研究的要求会有一些差异”,陈祖武介绍说:“会有更多的第三方供应商(主要是临床CRO机构)的进入,有更多的实验室(比如独立的医学影像中心)的介入,这会产生比较大的费用。”
创新药企早期普遍难以构建起完善的临床团队,因其牵涉到医学、统计等方方面面的人才。所以有些药企在药品的临床一期即采取外包策略,有的药企则在完成一期临床试验后,在更复杂的二期临床阶段开始采取外包策略。
2019年,中国药物临床试验的数量超过1600个,相比十年前不足100个的临床试验数量增长超过20倍。这其中,肿瘤药物的临床实验是占据了最大比例。不过,不管是恒瑞医药、百济神州等大药企自己组建临床研究团队,还是基石药业、信达医药等寻求第三方的CRO团队协同,都最终指向医院等临床机构的需求。
但中国的有临床试验资质的医院并不充足。
据《构建中国医药创新生态体系》报告的数据,国内临床试验中心数量由2015年的375家增长到2020年的1078家。但“多数中国临床研究机构的过往经验仍普遍不足。 在2019-2020年承担临床试验的1078家机构中,两年内承担超过20项临床研究的机构不到机构总数的30%。中国机构承担国际多中心临床试验的经验更为匮乏,仅有6%的机构在过去两年间承担超过20项国际多中心临床试验。”
高水平的学术带头人更加稀缺。
“我们的临床机构是增加的,但是金字塔尖的专家数量还是相对恒定。如果临床研究突然增长的话,这部分资源还是会紧缺,因为大家都会找最顶级的专家。以黑色素瘤临床研究为例,大家都想找北大肿瘤医院的朱军教授。”陈祖武表示。
一项肿瘤药物的临床试验,需要顶级的学术牵头人,以及与之配合的两三百科研、实验室等人员。快速增长的临床研究需求,以及稀缺的专家团队,导致肿瘤临床阶段的开支水涨船高。
“临床试验成本还是比美国低的,我觉得要低30%以上”,赵萍对第一财经记者表示,不过“成本一定是越来越高的”。
效率为王
除了研发阶段的成本压力,药企面临的另外一项压力是肿瘤药物价格的普药化趋势。
中国的医保政策目标是通过集中竞价谈判等措施来降低药品进入医院端的价格,从而构建一个更加普惠的医疗系统。不管医药企业喜欢与否,肿瘤药物近年来降价趋势明显,PD-1抑制剂领域尤其如此。
在百时美施贵宝的欧狄沃最初进入中国市场的时候,患者每年的治疗费用可达50万左右。在此后的数年时间里,中国的创新药企快速跟进,截至目前已经有五款本土企业研发的PD-1抑制剂上市。而今年8月获批上市的康方生物PD-1抑制剂安尼可,叠加患者救助项目的话,两年费用已可控制在4万元内。
肿瘤药物企业同时面临研发成本的刚性上升,医保控费的价格天花板。快速实现商业化,成为不二的选择。这也是为什么众多肿瘤药物优先选择一个小的适应症破冰。
国家政策规定,对治疗严重危及生命且尚无有效治疗手段疾病以及公共卫生方面等急需的药品医疗器械,临床试验早期、中期指标显示疗效并可预测其临床价值的,可附带条件批准上市。
据陈祖武介绍,为了加快肿瘤药物上市,不少药企采用的策略是先找一些“无药可用”的小瘤种,只做仅有几十例受试者的二期试验,在效果验证后容易获批上市。药企此后再通过上市后研究,将药品使用范围扩展至患者更多的大适应症,从而增加患者受益,延长产品生命周期。
目前国内上市的几款PD-1药物均是以小适应症获批,实现初步商业化后再寻求上市后的临床研究。据药研社统计的中国临床试验登记注册中心数据,2019年登记的肿瘤领域上市后临床研究共有1290项,2020年登记有2072项,年增长率高达60.6%。
肿瘤药物获批后快速推向医生和患者,同样至关重要。赵萍对记者表示:“对于大多数企业来说,过去的人海战术行不通了,我看到很多公司都在提升效率的过程中,这是企业未来考虑的,商业模式上要更加有效。”
中国肿瘤药物市场的创新生态正在形成,竞争的强度也在提升。赵萍认为,“肿瘤药物的全产业链在加速过程中”。如果研发缺乏效率,或者商业化缺乏效率,每个环节都可能把一家创新公司拖垮。
相关推荐
-
 安徽财政多措并举促进就业局势持续稳定
安徽财政多措并举促进就业局势持续稳定
-
 吉林省组织开展就业援助月专项活动 重点帮扶这三类
吉林省组织开展就业援助月专项活动 重点帮扶这三类
-
 河北沧州:2021年发放创业担保贷款48138万元 带动
河北沧州:2021年发放创业担保贷款48138万元 带动
-
 晋中创新性构建三个“1+N”体系 服务高校师生创新
晋中创新性构建三个“1+N”体系 服务高校师生创新
-
 1950人入围!首批“浙江青年工匠”入选名单公布
1950人入围!首批“浙江青年工匠”入选名单公布
-
 滨州市公共就业服务平台上线 开启“灵活用工和共享
滨州市公共就业服务平台上线 开启“灵活用工和共享
-
 2022年给职场人的十大建议
2022年给职场人的十大建议
-
 今年山东省智能建造与建筑工业化职业技能竞赛在济南
今年山东省智能建造与建筑工业化职业技能竞赛在济南
-
 2021年“创业常州”讲堂活动圆满落下帷幕
2021年“创业常州”讲堂活动圆满落下帷幕
-
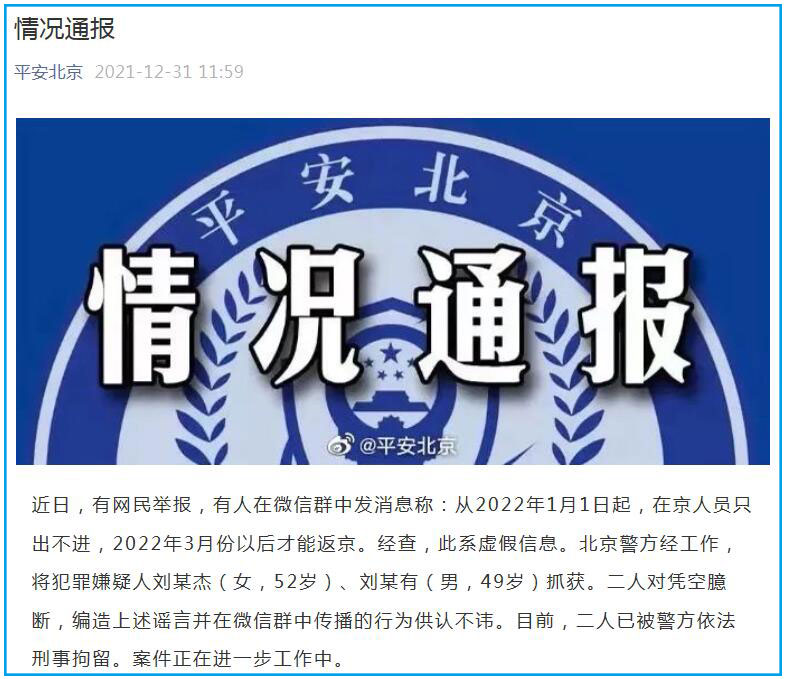 造谣“在京人员只出不进 3月才能返京” 二人被北
造谣“在京人员只出不进 3月才能返京” 二人被北
-
 浙江上虞发布:离虞需携带48小时核酸检测报告!查验
浙江上虞发布:离虞需携带48小时核酸检测报告!查验
-
 铁路货运电子商务平台近日升级上线 24小时网上办理
铁路货运电子商务平台近日升级上线 24小时网上办理
-
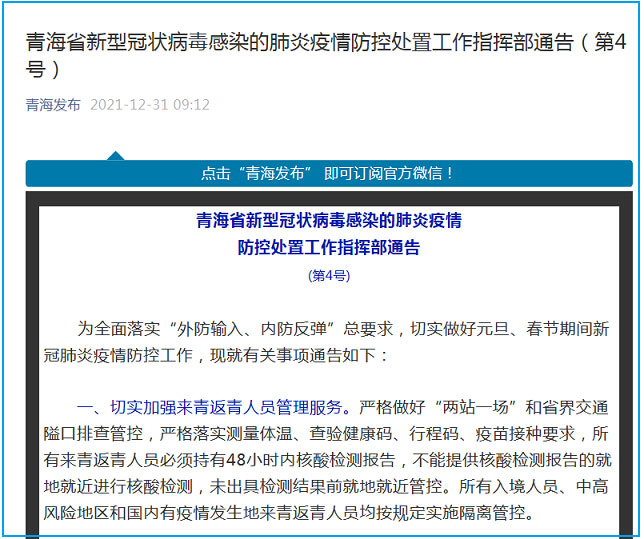 青海:切实加强来青返青人员管理服务 从严控制人员
青海:切实加强来青返青人员管理服务 从严控制人员
-
 12月31日气象报告:元旦假期全国大部天气晴好
12月31日气象报告:元旦假期全国大部天气晴好
-
 各种补贴 力度加大!北京扶持养老驿站办法明年实施
各种补贴 力度加大!北京扶持养老驿站办法明年实施
-
 黑龙江:拟认定省第二批制造业“隐形冠军”企业名单
黑龙江:拟认定省第二批制造业“隐形冠军”企业名单
-
 齐齐哈尔市“引才工作”再添佳绩 为这座城市注入新
齐齐哈尔市“引才工作”再添佳绩 为这座城市注入新
-
 新增入统规上企业每户奖50万!哈尔滨支持制造业发展
新增入统规上企业每户奖50万!哈尔滨支持制造业发展
-
 牡丹江市打造“诚信龙江”品牌 “码上诚信”企业占
牡丹江市打造“诚信龙江”品牌 “码上诚信”企业占
-
 今年黑龙江粮食总产量达到1573.54亿斤 粮食总产量
今年黑龙江粮食总产量达到1573.54亿斤 粮食总产量